
产品注册/备案是医疗器械生命周期中的重要环节,产品注册证是医疗器械产品的上市销售许可证明,只有成功获得注册证的医疗器械产品,才能在中国境内合法销售。什么样的产品需要注册/备案?
医疗器械分类管理方式及监管部门有哪些?
第Ⅰ、Ⅱ、Ⅲ类医疗器械备案流程及材料要求?
医疗器械注册/备案相关信息查询常用网站有哪些?
本文所介绍的三类医疗器械注册/备案的流程与申报材料要求,依据的是2021年6月1日开始执行的《医疗器械监督管理条例》,及配套的《医疗器械注册与备案管理办法》及《医疗器械注册申报资料要求和批准证明文件格式》等。
《医疗器械监督管理条例》原文官网链接:
https://www.nmpa.gov.cn/xxgk/fgwj/flxzhfg/20210319202057136.html
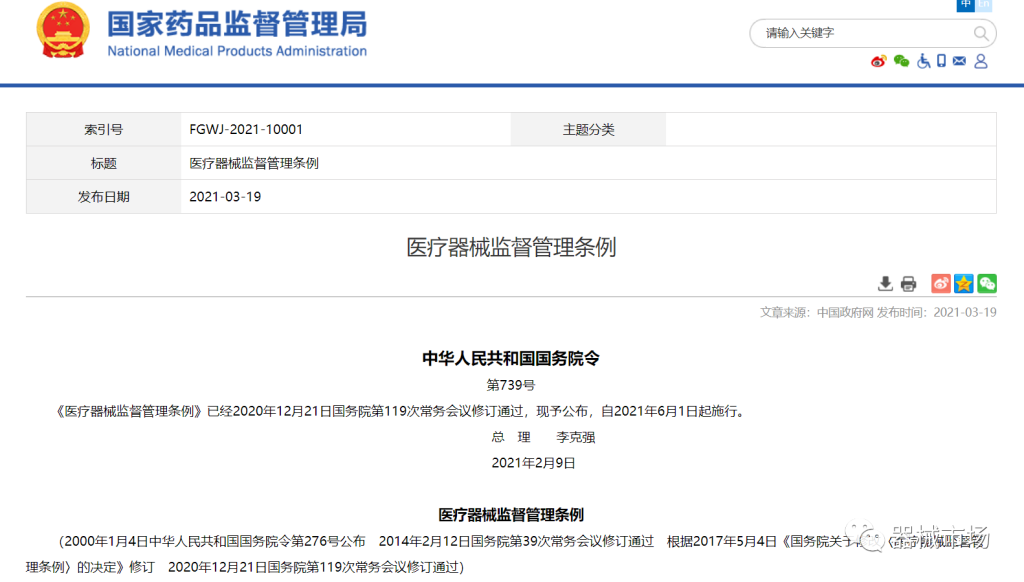
医疗器械分类管理方式及监管部门
【什么样的产品需要进行注册/备案】《医疗器械监督管理条例》第一百零三条,明确进行了定义:
医疗器械,是指直接或者间接用于人体的仪器、设备、器具、体外诊断试剂及校准物、材料以及其他类似或者相关的物品,包括所需要的计算机软件;其效用主要通过物理等方式获得,不是通过药理学、免疫学或者代谢的方式获得,或者虽然有这些方式参与但是只起辅助作用;其目的是:
(一)疾病的诊断、预防、监护、治疗或者缓解;
(二)损伤的诊断、监护、治疗、缓解或者功能补偿;
(三)生理结构或者生理过程的检验、替代、调节或者支持;
(四)生命的支持或者维持;
(五)妊娠控制;
(六)通过对来自人体的样本进行检查,为医疗或者诊断目的提供信息。
如果无法判断一个产品是否具有医疗用途,可申请分类界定,请监管部门帮忙判断。
从产品组成上看,如果一个器械产品含有药品成分,药理作用再叠加器械本身的物理作用 → 实现医疗用途。这种情况下,请问,这个产品是属于医疗器械还是药品?为什么一定要弄清这个问题呢?因为监管机构不同,器械的监管机构是CMDE(医疗器械技术审评中心),而药品的监管机构是CDE(药品审评中心),完全不同的两个部门。当然也存在两个部门联合审评的情况,即CMDE审核器械部分的相关资料,CDE审核药品相关的资料,最后再整合在一起给意见。那就是非常复杂的产品了,很少会遇到。
如果不知道如何判断产品到底是器械为主还是药品为主,可申请属性界定,请监管部门帮忙判断。
【医疗器械的管理类别】我国根据医疗器械的风险等级,将医疗器械产品分为以下三大类:I类 :风险程度低,实行常规管理可以保证其安全、有效的医疗器械。 II类 :具有中度风险,需要严格控制管理以保证其安全、有效的医疗器械。III类:具有较高风险,需要采取特别措施严格控制管理以保证其安全、有效的医疗器械。
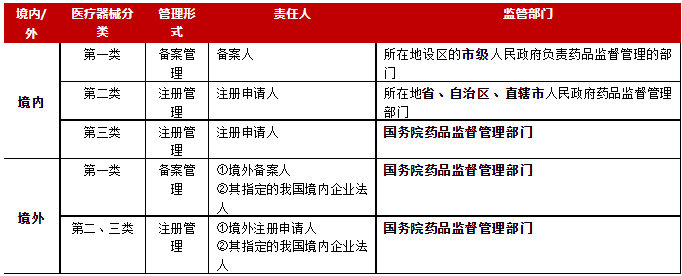
第一类医疗器械备案流程及材料要求
(一)第一类医疗器械备案流程与时限

1.第一类医疗器械备案表;2. 关联文件;3.产品技术要求;4.产品检验报告;5.产品说明书及最小销售单元标签设计样稿;6.生产制造信息;7.符合性声明;8.经办人授权证明和经办人身份证复印件;9.变化情况说明及相关证明文件;10.补发第一类医疗器械备案编号告知书情况说明;11.第一类医疗器械备案编号告知书遗失后备案人向原备案部门的报告;12.取消备案说明及第一类医疗器械备案编号告知书。A、首次备案:1-7。B、变更备案:9、8。(变更企业名称、地址、产品技术要求、规格型号、第一类医疗器械备案信息表登载其他内容等要素时,需要提交企业变更后的营业执照、房屋产权证明或者房屋租赁合同、变更后的产品技术要求、规格型号等相关证明)C、补发凭证:10、8。【备注】补发的第一类医疗器械备案编号告知书,发放日期为补发当日。D、取消备案:12、8。03
第二类医疗器械注册流程及材料要求
(一)第二类医疗器械注册流程及时限
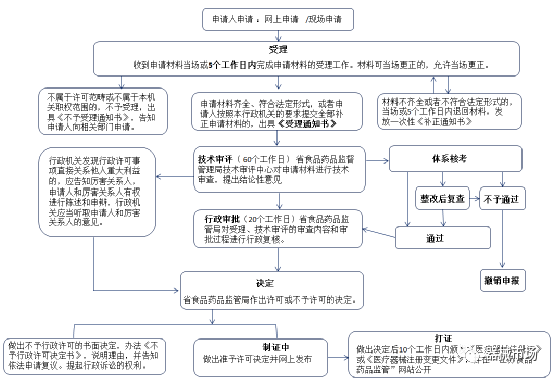

04
第三类医疗器械注册流程及材料要求
(一)注册流程与时限
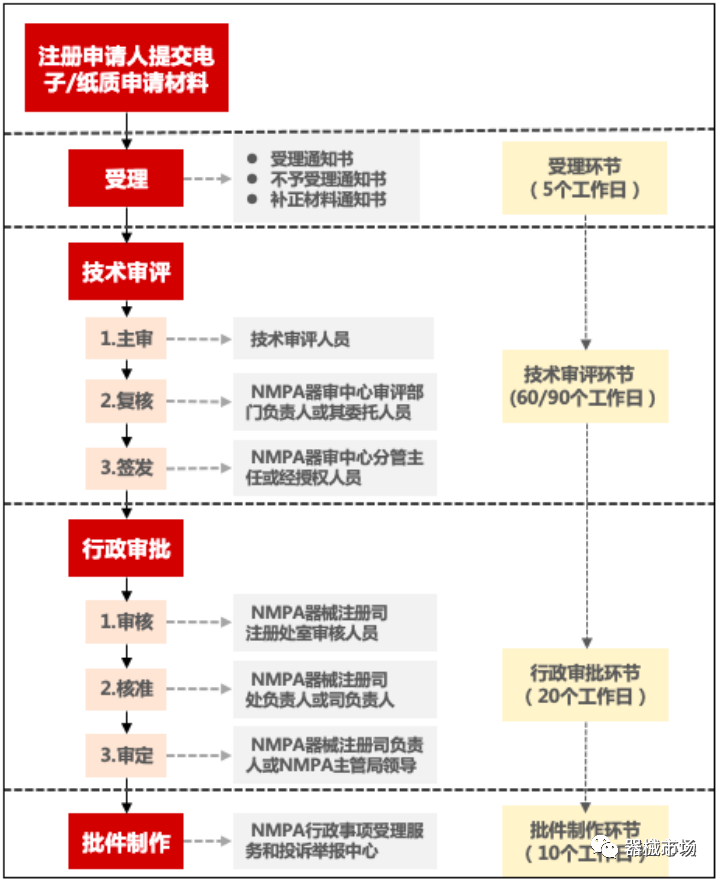
二)第三类医疗器械注册申请材料
第三类医疗器械注册和监管最为严格,因而申请材料也较多,分为境内和境外第三类医疗器械注册申报资料。因篇幅有限,具体申报资料内容不在此展示,感兴趣的朋友可联系我们 150 1316 6558 (微信同号)